日本のラグ問題をアップデートする 〜ドラッグ編〜
日常臨床の中で、日本が抱える「ラグ問題」について考えたことのある先生は多くいらっしゃると思います。私も、「海外では承認されているあの薬、あのデバイスが今使えたら、もっとこの患者さんはよくなるのに」と思ったことは何度とあります。ドイツに来てから、研究所と医薬品企業との合同会議に立ち会ったり、PMDAのスタッフの方とお話をしたりする機会があり、この「ラグ問題」について、多角的に捉え理解を深めるきっかけになっています。
そこで今回、この「ラグ問題」をドラッグ編とデバイス編の2編に分けて、レビューしてみたいと思います。これまでも多くの議論がある題材ですが、できる限り最新の情報を拾い、要点を簡潔にまとめます。
それではまず、ドラッグ編です。
<ドラッグラグはどうして生じるのか>
ラグはどうすれば減らすことができるか。ということを考える前には当然、ラグが何故生じているのかを知る必要があります。ここからスタートします。
ドラッグラグは、製薬企業から規制当局(日本の場合はPMDA)への承認申請の遅れ、すなわち開発・治験期間による「開発ラグ」と、規制当局申請後の審査の遅れによる「審査ラグ」の総和として発生します(図1, 文献1)。
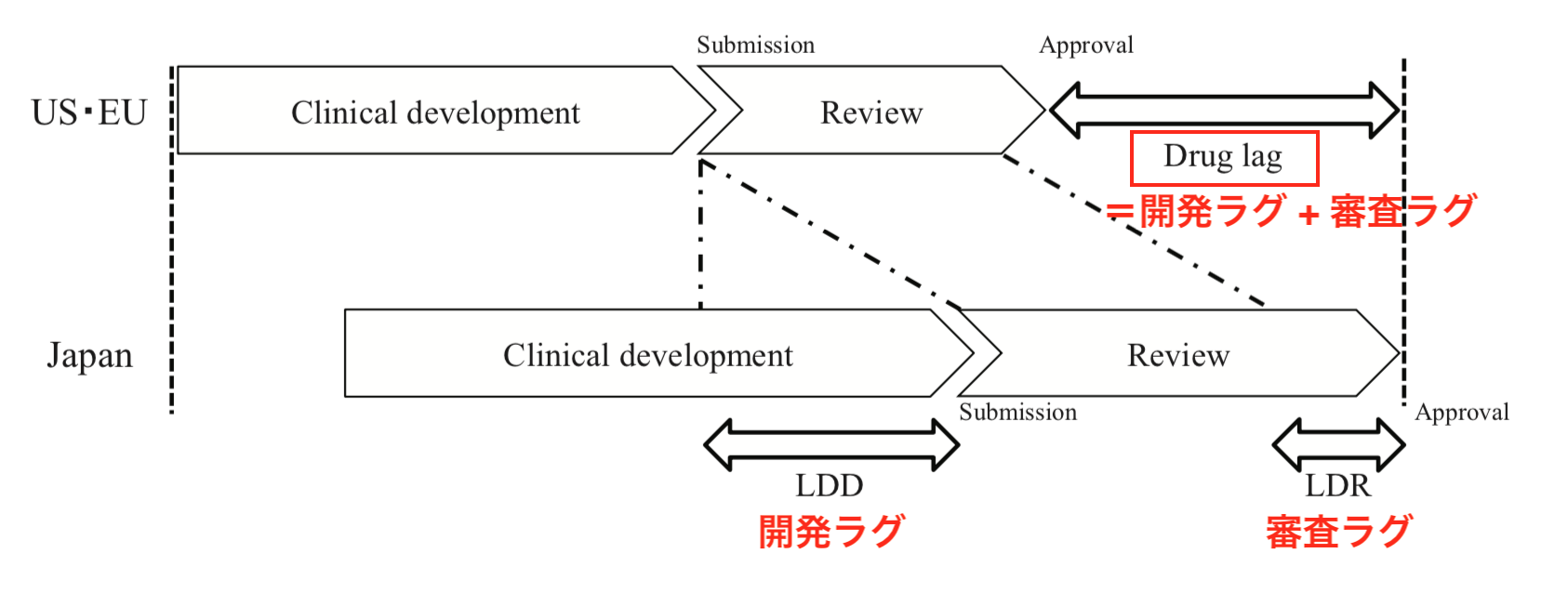
図1. ドラッグラグが起こる仕組み
これらの年次推移を見てみると、事実、近年ドラッグラグは徐々に解消されつつあり、その主な理由が審査ラグの劇的な減少であることがはっきりと見て取れます(図2)。さらに各地域の規制当局の審査期間を比べた場合、PMDAの審査は2011年以降、最短の米国FDAと肩を並べる程度まで短くなっています(図3, 文献2)。
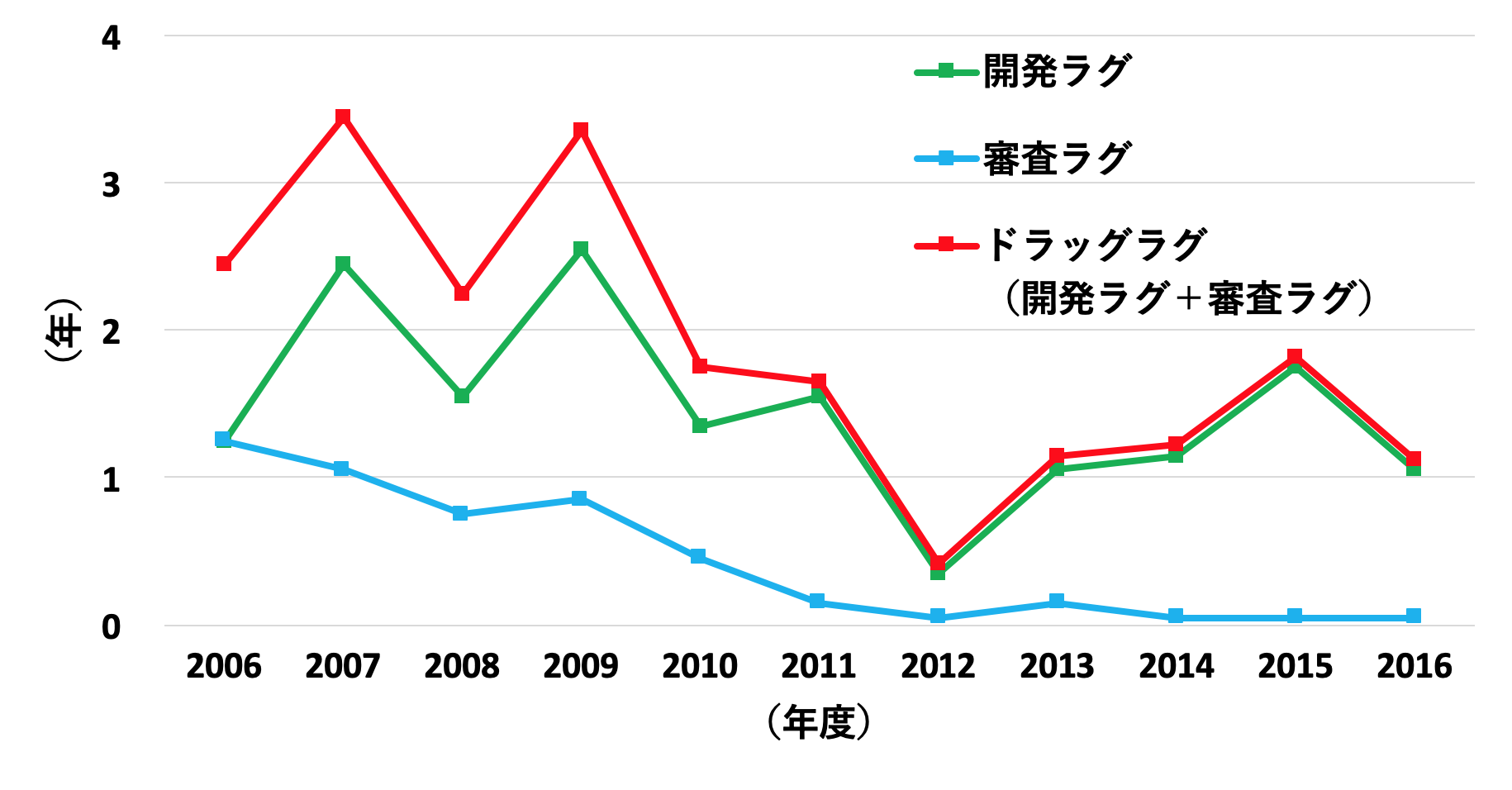
図2. ドラッグラグの年次推移 (PMDA発行資料を基に筆者が作成)
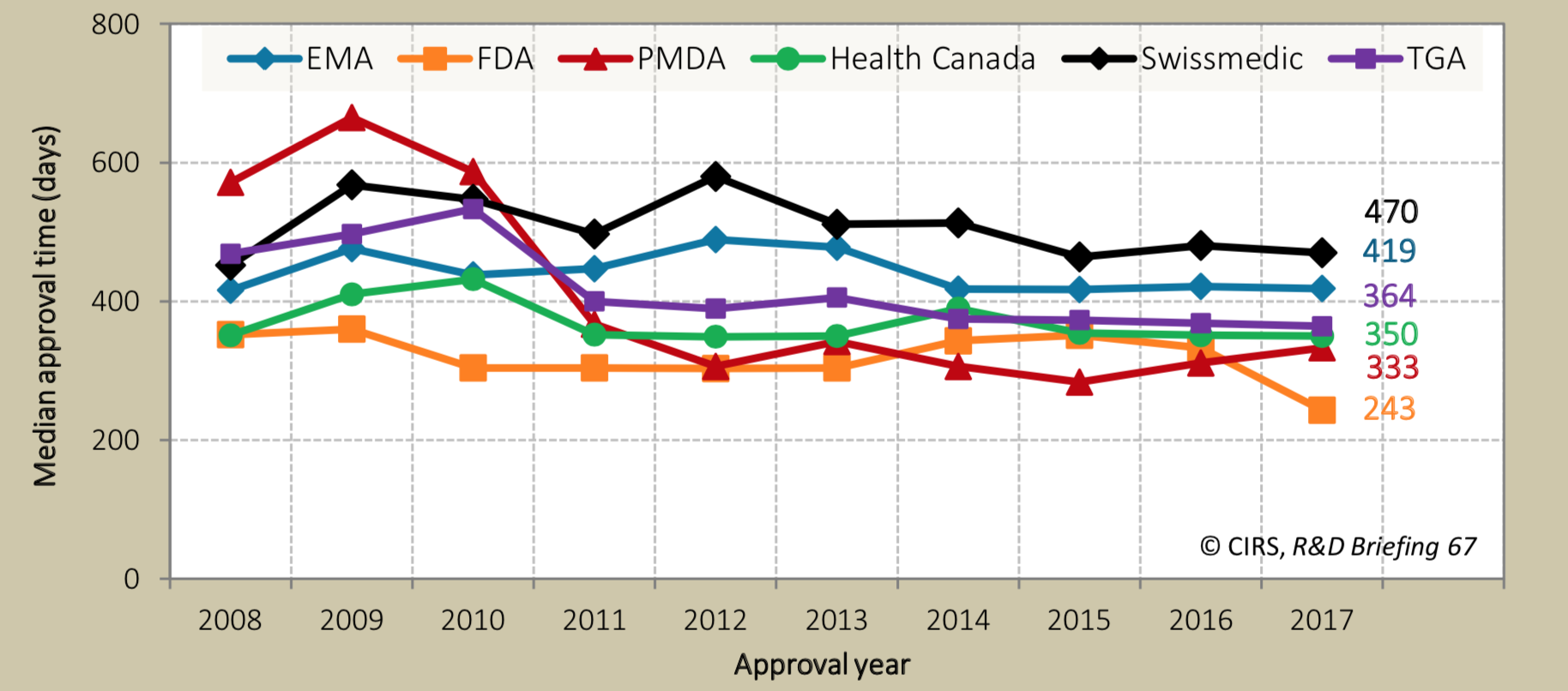
図3. 規制当局の新薬承認審査期間の地域別年次推移 (EMA, FDA, PMDA, Health Canada, Swissmedic, TGAは、それぞれEU, 米国, 日本, カナダ, スイス, オーストラリアの規制当局)
この審査ラグの減少には、2000年代から施策としてPMDAの審査スタッフが増員されてきたことが大きく寄与しています(図4)。
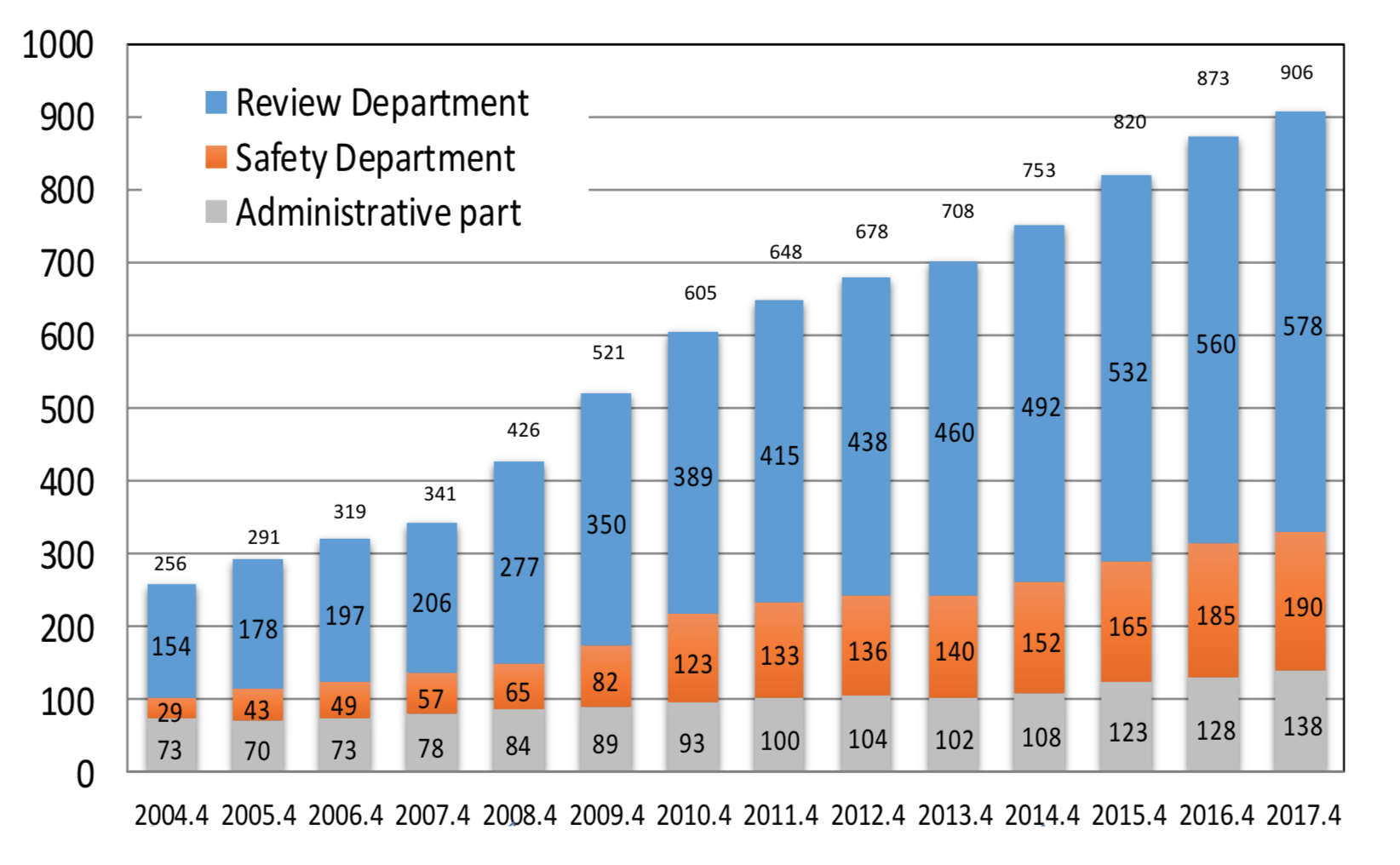
図4. PMDAスタッフ数の年次推移 (PMDA発行資料より)
これらのデータから、PMDAの尽力により審査ラグが十分に解消された現在、ドラッグラグのさらなる解消を目指すためには開発ラグを短縮する必要がある、ということがわかります。
<開発ラグはどうして生じるのか、どのように対応するべきか>
それではこの開発ラグはどのような理由で生じているのでしょうか。下記にその要因を列挙し、それぞれの対応策について考えてみたいと思います。
① 日本人対象者を含む治験実施による遅れ
PMDAは新薬の承認申請にあたり、先行する欧米の臨床試験で安全性・有効性が示されている医薬品に関しても、原則的に日本人を対象としたデータの提出を求めます。ここで日本国内で別途治験を組むことになれば、相当の時間を要し、結果的に大きなラグを生みます。
このステップにおいて、先行する他地域との足並みを揃える手段として、国際共同治験に日本人症例を組み入れることの有用性が以前から指摘されています。実際最近では、日本が参加する国際共同治験が増えてきていますが(図5, 文献3 )、日本では例えば治験の専門人材(治験コーディネーター、データマネージャーなど)が十分には配置されていないなど、治験を支える環境整備には改善の余地が残されています(文献1)。また、PMDAは承認申請前の治験相談として、製薬企業に対して適切な試験デザインやサンプルサイズに関する助言を提供していますが、企業は新薬開発のなるべく早い段階からこれを利用して開発計画を立案する必要があります。なお、「先駆けパッケージング戦略」や「条件付早期承認制度」など、治験の実施を必要としない承認スキームも存在しており、これらに関しては次回デバイス編でもう少し詳しく取り上げたいと思います。
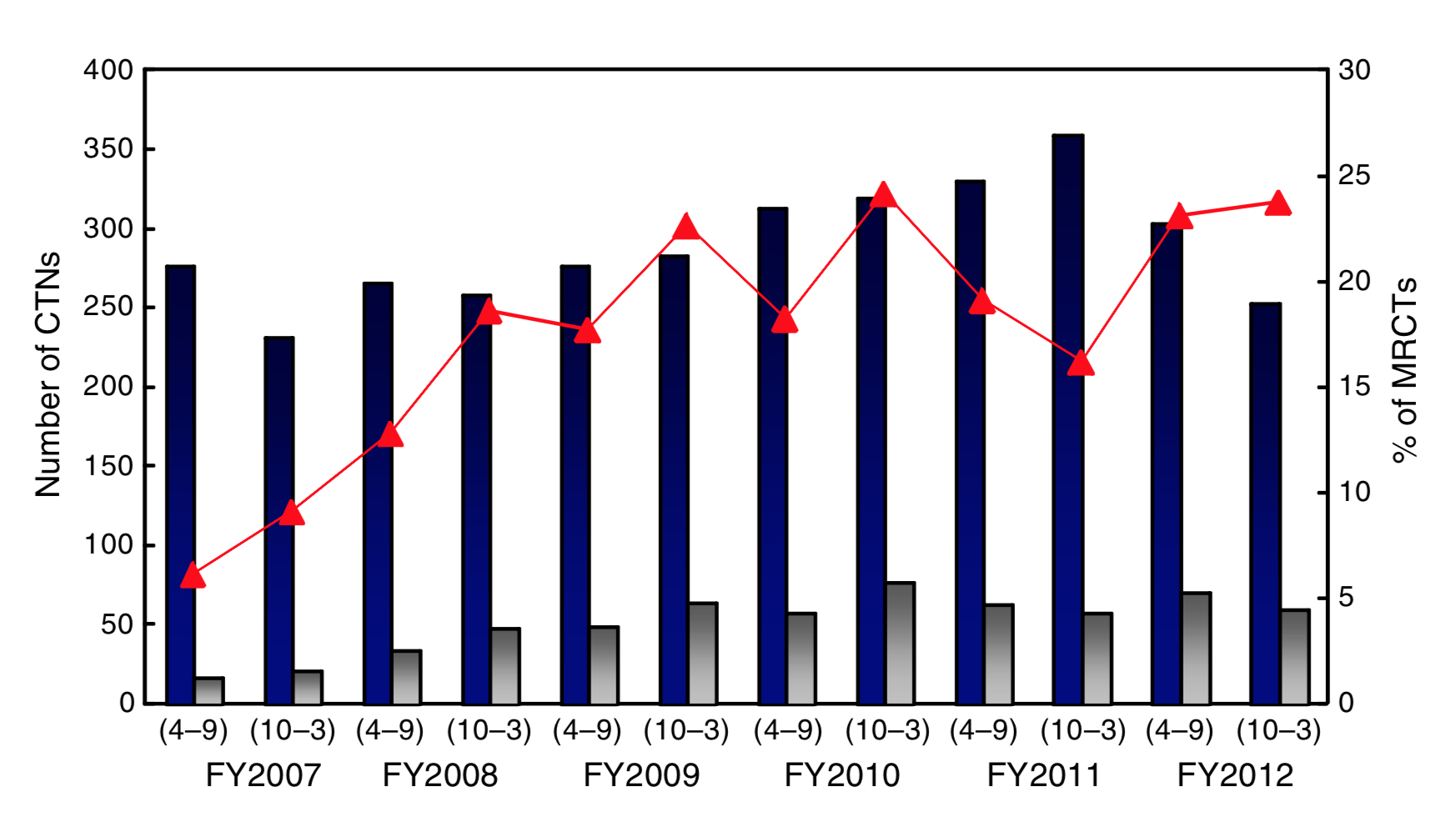
図5. 日本が参加する国際共同治験の推移 (濃紺バー: PMDAに届出される全治験数, 灰色バー: そのうちの国際共同治験数, 赤折線: 後者の割合, CTNs: clinical trial notifications, MRCTs: multi-regional clinical trials)
② 海外企業によって開発される医薬品
一般的に、製薬企業の本社のある国・地域で優先的に当該医薬品の開発が進められる傾向があると言われており、日本国内で開発された医薬品は海外企業によって開発された医薬品と比べ、開発ラグが有意に短い(0日 vs. 954日)という報告もあります(文献1)。
よって、国内での医薬品開発を活発化させる必要があり、このために産官学が一体となることが一層重要となります。この動きを推進するべく、2015年には、医療研究開発における基礎から実用化までの過程をシームレスに支援するAMEDが発足し、厚生労働省からは医薬品産業強化総合戦略が打ち立てられています。これらによって開発ラグに及ぼされる効果が見えるのは、もう少し先のことになりそうです。
③ PMDA特有の審査基準
元来の保守的な国民性に加え、サリドマイド事件や血液製剤による薬害C型肝炎など、薬害を経験するたびに当時の規制当局のあり方が批判されてきた歴史的背景もあって、日本の新薬承認プロセスは米国・EUと比べ、安全性により重点が置かれています(文献4)。この慎重なスタンスは確かにラグにつながりますが、一方で、ラグのおかげで、重大な安全上の問題が他地域で検出された場合でもその被害を避けることができる、という利点が併存しているのも事実です。実際に、ラグが長い薬剤の方が、市販後に通達される注意喚起の頻度が少ない、という報告もあります(文献5)。ただ現在の趨勢としては、有効な治療薬をいち早く患者さんに届ける、というラグ解消に伴う利益の方がより重要とみなされています。
少し話が逸れましたが、その他にも、PMDAの審査では米国FDAやEUのEMAと異なる項目が要求され、これを準備する作業だけでも時間がかかります。今後は、各地域の規制当局間で連携を図り開発途中の医薬品についての情報交換を活発に行うことや、各々の申請手続きの親和性を高めることなども、課題になってくるでしょう(文献6)。
④ 市場としての魅力の低さ
日本の医薬品市場は、国民皆保険制度のもと厚生労働省によって薬価が一律に低く抑えられているため、製薬企業が多額の開発コストに見合う売上を回収しにくい、という性質を持ちます。そのため、企業にとってより経済的インセンティブの働く他地域での承認申請が優先されることになり、結果的にラグが生まれます(文献4)。さらに、2018年に行われた薬価制度の抜本改革では、新薬創出加算の対象が減らされ、主に薬価の引下げが行われる薬価改定を隔年から毎年に増やすことが決められました。これらは追い打ちをかけるように、新薬開発を目指す製薬企業の国内事業予見性を低下させています。
ともすれば②に述べた国内の医薬品開発を推進する施策と矛盾するようにも受け取れる、この市場としての側面は、日本独自の皆保険制度や逼迫する医療財政と直接的にリンクしており、開発ラグ解消を目指すにあたり大きな壁として立ちはだかっています。
<引用文献>
2. Centre for Innovation in Regulatory Science. R&D Briefing 67. 2018. https://www.cirsci.org/wp-content/uploads/2018/05/CIRS-RD-Briefing-67-04052018_FINAL.pdf
3. Honig PK. Clin. Pharmacol. Ther. 2014;95:467–469.
4. Tanimoto T. Drug Des Devel Ther. 2015;9:1877-1888.
5. Yamada T, et al. Ann Pharmacother. 2010;44:1976–1985.
6. Kondo H, et al. Ther Innovation Regulatory Sci. 2018;52;214-219.
ドラッグラグとデバイスラグは内包する課題に共通点も多く存在し、今回記述したことはある程度デバイスラグにも当てはまります。次回のデバイス編ではデバイスラグ特有のことにフォーカスしたレビューをお届けし、最後に2編をまとめた個人的な感想を述べたいと思います。
Warning: Trying to access array offset on null in /home/c2843648/public_html/sunrise-lab.net/wp-content/themes/betheme-child/includes/content-single.php on line 278